|
|
|
|
In sensul cel mai larg dinamica moleculara se ocupa cu miscarea moleculara inerenta tuturor proceselor chimice; vibratia atomilor in molecula, reactiile chimice, interactia substrat-receptor, si multe alte procese complexe sunt asociate cu diferitele forme de miscare intra- si intermoleculare.
In timp ce mecanismul unui proces chimic este descris de cinetica chimica, energetica acestor procese reprezinta obiectul termodinamicii. Cu alte cuvinte cinetica chimica este aceea care prezinta succesiunea si viteza evenimentelor care apar la transformarea sau trecerea moleculelor dintr-o stare in alta, iar termodinamica dicteaza relatiile energetice intre diferitele stari moleculare posibile.
Un sistem molecular, aflat la o anumita temperatura, este caracterizat cu acuratete de miscarea sa. Dinamica moleculara simuleaza aceasta miscare in spatiul fazelor (spatiul definit prin pozitia si vitezele atomilor), prin integrarea numerica a ecuatiilor de miscare date de Newton. Mecanica moleculara modifica gradele de libertate intramoleculare intr-o maniera asemanatoare cu cea folosita in minimizarea energiei: etapele individuale in minimizarea energiei constau in cautarea directiei de coborare catre un minim; etapele individuale in simularile dinamicii moleculare constau in calcularea noilor pozitii ri+1 si viteze vi+1 ale atomilor, pe baza celor curente (ri, vi) si a modificarilor in timp.
Viteza si directia de deplasare este determinata de fortele pe care atomii sistemului le exercita unul asupra celuilalt, forte descrise de ecuatiile lui Newton.
In practica, atomilor li se asociaza viteze initiale, corespunzatoare energiei cinetice pe care sistemul o poseda, energie determinata la randul ei de temperatura dorita pentru simularea respectiva. Aceasta temperatura este atinsa printr-o incalzire lenta a sistemului, plecand de la zero absolut, urmata de o perioada de atingere a echilibrului.
Intr-o simulare de dinamica moleculara mai intai se calculeaza fortele care actioneaza asupra fiecarui atom, prin variatia energiei potentiale ca urmare a variatiei pozitiei cu o cantitate infinitezimala dri; cu alte cuvinte, forta care se modifica in timp, este egala cu gradientul cu semn schimbat al energiei potentiale:
![]() (III.1)
(III.1)
unde E reprezinta energia potentiala, iar ri pozitia atomului i.
Energia poate fi calculata fie folosind mecanica moleculara, fie metodele mecanicii cuantice. Mecanica moleculara este limitata la acele procese care nu afecteaza drastic structura electronica a sistemului, cum ar fi formarea sau ruperea unei legaturi chimice.
Cunoscand fortele care actioneaza asupra atomilor si masa acestora mi, se pot determina pozitiile lor de-a lungul unei serii de intervale de timp extrem de scurte de ordinul femtosecundelor (10-15 s). Schimbarile structurale rezultate reprezinta asa numita traiectorie in timp a sistemului molecular.
Ecuatia (III.1) poate fi scrisa sub forma:
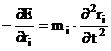 (III.2)
(III.2)
In practica traiectoria nu se obtine prin rezolvarea ecuatiei (III.2) datorita lipsei unei solutii analitice. De aceea, cunoscand fortele si masele se determina acceleratiile ai. Vitezele si pozitiile atomilor se obtin prin integrarea acceleratiilor, respectiv a vitezelor in raport cu timpul, folosind relatiile:
![]() (III.3)
(III.3)
Algoritmul folosit la integrare este cel al sariturii de broasca":
traiectoria
intre doua stari este impartita in intervale de timp
extrem de mici ![]()
odata calculata acceleratia la momentul t, ai(t), se determina viteza la jumatatea intervalului:
![]() (III.4)
(III.4)
noua
pozitie a atomilor dupa intervalul ![]() , se calculeaza
folosind viteza la mijlocul intervalului:
, se calculeaza
folosind viteza la mijlocul intervalului:
![]() (III.5)
(III.5)
In
felul acesta in timp ce pozitiile si acceleratiile sunt
determinate dupa intervale de timp intregi ![]() , vitezele se determina la jumatati de
intervale, sarindu-se peste valorile intregi. Din aceasta cauza
valorile energiei totale sunt aproximative.
, vitezele se determina la jumatati de
intervale, sarindu-se peste valorile intregi. Din aceasta cauza
valorile energiei totale sunt aproximative.
Utilizand intervale de timp extrem de scurte de 0,5 - 1 femtosecunde (fs), in cazul folosirii tuturor atomilor si de 1 - 2 fs pentru modelul atomului unit, se realizeaza o integrare adecvata pentru miscarile de inalta frecventa ale sistemului molecular (de exemplu vibratiile de valenta presupun perioade de ordinul picosecundelor).
Aceasta constituie, in acelasi timp si un dezavantaj al simularilor dinamicii moleculare, deoarece lungimea traiectoriei, calculata intr-un timp rezonabil, este cu cateva ordine de marime mai scurta decat orice proces chimic si decat foarte multe procese fizice, a caror durata poate fi de ordinul nanosecundelor sau chiar mai mare. De aceea prin dinamica moleculara pot fi studiate practic numai acele procese si proprietati, care dureaza mai putin decat durata simularii, cum ar fi fluctuatiile in energie sau schimbarea pozitiei atomilor, dar nu procese de durata, ca de exemplu plierea proteinelor.
In lipsa unui termostat neexistand nici o sursa externa , sau rezervor de energie, energia totala a sistemului este constanta. Intr-un ansamblu microcanonic (numarul particulelor, volumul si energia constante), teoretic, schimbarile in energia cinetica ar trebui sa fie de aceeasi marime, dar de semn opus cu cele ale energiei potentiale, astfel incat o simulare de dinamica moleculara sa poata fi facuta in conditiile de conservare a energiei; practic conservarea energiei este dificil de atins datorita limitarilor atat de capacitate cat si de viteza ale tehnicii de calcul. Aceste limitari impun o precizie finita si un calcul aproximativ al energiei, datorita in primul rand trunchierii la o distanta finita a interactiilor atomilor nelegati.
O
alta posibilitate este de a efectua simularea dinamicii moleculare in
conditii de temperatura constanta prin adaugarea unui
termen cu ajutorul caruia se ajusteaza vitezele atomilor
mentinand sistemul la temperatura dorita; in timpul unei astfel de
simulari vitezele sunt ajustate prin scalare cu un factor ![]() dupa fiecare
interval de timp
dupa fiecare
interval de timp ![]() ; sistemul este cuplat cu o baie de incalzire la
temperatura de simulare T0 si cu un timp de relaxare
; sistemul este cuplat cu o baie de incalzire la
temperatura de simulare T0 si cu un timp de relaxare ![]() . In aceste
conditii factorul de scalare
. In aceste
conditii factorul de scalare ![]() este:
este:
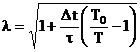 (III.6)
(III.6)
Pentru
o traiectorie stabila se utilizeaza o constanta de relaxare ![]() mai mare decat 0,1 ps; o
constanta de 0,01 ps este prea mica si creeaza distorsiuni
in simulare.
mai mare decat 0,1 ps; o
constanta de 0,01 ps este prea mica si creeaza distorsiuni
in simulare.