|
|
|
|
Introducere
Prionii sunt particule infectioase care cauzeaza un grup de maladii neurodegenerative caracterizate prin degenerarea sistemului nervos central. Sunt lipsiti de orice tip de acid nucleic si pot induce o serie de maladii atat la om cat si la animale. La om, cele mai cunoscute sunt: maladia Creutzfeld-Jacob (CJD), sindromul Gerstmann-Straussler-Scheinker (GSS), insomnia fatala familiala (FFI), maladia Kuru si sindromul Alpers (intalnit numai la copii), iar la animale: scrapie (la oi si capre), encefalopatia spongiforma bovina (BSE) sau "boala vacilor nebune", encefalopatia spongiforma a felinelor, a cervidelor, etc.
Procesul care declanseaza boala este reprezentat de conversia unei proteine normale, sintetizata in mod natural in creierul tuturor mamiferelor (PrPc), intr-una mutanta, anormala (PrPSc). In functie de factorul care induce conversia PrPc in PrPSc, natura acestor afectiuni poate fi genetica (in cazul unei mutatii punctiforme a genei pentru PrP, transmisa autozomal dominant), infectioasa (ca urmare a consumului alimentelor contaminate, a folosirii unor instrumente chirurgicale nesterile, injectarii unor hormoni derivati din hipofiza prelevata de la cadavre), sau sporadica (datorata unor mutatii spontane ale genei PrP, care intensifica rata conversiei proteinei prionice, cat si unor factori necunoscuti).
Maladiile prionice reprezinta un grup heterogen de disfunctii neurodegenerative cunoscute ca encefalopatii spongiforme transmisibile (TSE), ce au in comun leziuni cerebrale, produse prin degenerescenta vacuolara a neuronilor. Post-mortem se constata un aspect spongios al creierului, aspect datorat prezentei vacuolelor care se gasesc in special la nivelul substantei cenusii, corespunzator corpilor neuronali.
Perioada de latenta a acestor maladii este variabila (luni sau chiar zeci de ani). Odata instalata, boala evolueaza spre debilitate mintala, dementa, pierderea controlului motor, imobilizare etc.
Interesul crescut fata de aceste afectiuni este datorat unor particularitati legate de structura, modul de transmitere si patologia indusa, intra sau interspecific, de catre prion.
Toate aceste maladii sunt asociate cu alterarea conformationala determinata de conversia formei normale a proteinei prionice, sensibila la proteaza (PrPC), la o forma rezistenta la proteaza (PrPSc), marker specific TSE-urilor; aceasta modificare are loc doar dupa ce proteina prionica a fost ancorata la nivelul membranei celulare.
Atunci cand la nivelul genei care codifica proteina prionica normala, au loc mutatii punctiforme, se realizeaza sinteza unei izoforme anormale, denumita, dupa numele bolii la care a fost studiat "PrP scrapie" sau PrPSc. Aceasta proteina scrapie mai este cunoscuta si sub numele de prion. Conceptul de prion a fost introdus de Prusiner in urma studierii scrapiei la oi (encefalopatia spongiforma a ovinelor). Prionii reprezinta o categorie de agenti infectiosi, considerandu-se ca sunt un mister din punct de vedere biologic, deoarece, in structura lor intra doar o molecula proteica. Fiind lipsiti de genom, difera de toate categoriile de agenti infectiosi cunoscute pana in prezent, prin faptul ca nu contin nici un tip de acid nucleic (ADN sau ARN). Este interesant de relatat faptul ca prionii rezista la actiunea multor factori fizico-chimici, printre care amintim: formol 10% (timp de 28 de luni), caldura (rezista la fierbere timp de 3 ore), factori inhibitori ai acizilor nucleici, precum si la actiunea radiatiilor UV (activitate infectioasa 100%).
Prin analogie, forma normala a acestei proteine, prezenta in conditii normale in celula, a fost desemnata PrPC (proteina prionica ceulara).
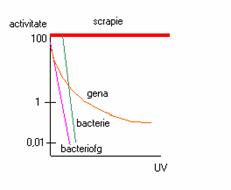
Figura 1. Reprezentarea activitatii infectioase a prionului si a organismelor cu grad diferit de organizare a ADN, sub actiunea UV.
In incercarea de a descifra cauza care sta la baza aparitiei acestor maladii, au fost formulate mai multe teorii. Imposibilitatea de a descoperi pe preparate provenite de la pacienti cu TSE, un agent infectios reprezentat de un acid nucleic, precum si rezistenta foarte mare a acestor maladii la tratamente, care de obicei distrug acidul nucleic, conduce la formularea ipotezei proteinei prionice infectante. Aceasta ipoteza a fost postulata in 1982, de catre Stanley Prusiner care sustine etiologia proteica a TSE, si presupune ca PrPSc ar actiona ca matrita in conversia izoformei PrPC normale, in PrPSc asociata cu boala. Astfel, in aceasta viziune, proteina prionica se autopropaga prin contactul cu proteina normala, pe care o determina intr-un anume fel sa adopte o conformatie diferita fata de cea initiala, luand forma prionica. Aceste modificari se propaga in lant, astfel incat noile particule infectioase modifica in continuare alte proteine prionice normale cu care vin in contact. S-a demonstrat ca in absenta acestei proteine normale, PrPSc este incapabila de a determina aparitia bolii. Una din lacunele acestei ipoteze este faptul ca nu poate explica cum o molecula proteica poate determina proprietati biologice diferite precum si diferite forme de TSE. S-ar parea, totusi, ca diferitele forme de TSE ar fi determinate de diferite conformatii si/sau de nivelul diferit de glicozilare al proteinei prionice.
O alta ipoteza incrimineaza un agent infectios asemanator virusurilor, numit virino, ale carui copii ar permite dezvoltarea diferitelor variante ale bolii neurodegenerative si, ocazional, schimbarea caracteristicilor prin mutatie. Acesta este un agent infectios subviral alcatuit dintr-o molecula mica de acid nucleic, asociat cu o proteina codificata de celula gazda. In aceste conditii se pare ca virino este asociat cu izoforma scrapie a proteinei prionice.
Totusi, ipoteza cea mai vehiculata este ca PrPSc ar fi ea insasi principalul si chiar singurul component al agentului infectios. Aceasta ipoteza, "protein only", propusa de Griffith in anul 1967, arata ca propagarea prionilor ar avea loc prin intermediul PrPSc care se replica cu inalta fidelitate prin recrutarea PrP endogene. In absenta acestei forme, s-a aratat ca PrPSc este incapabila de a se replica si de a induce boala. Pentru a fi infectioasa, PrPSc trebuie sa gaseasca in celula infectata molecule identice din punct de vedere chimic, pentru a le modifica structura.
Structura PrPC
Proteina prionica celulara este exprimata la nivelul sistemului nervos central si periferic, in tesutul limfatic si la nivelul jonctiunilor neuromusculare, ca o proteina situata la suprafata celulei, fiind ancorata de membrana celulara prin intermediul unei ancore glicolipidice. In mod normal, aceasta forma este produsa in creierul tuturor mamiferelor si este inofensiva, insa, in anumite conditii poate adopta o forma alterata, care reprezinta agentul infectios implicat in dezvoltarea TSE. Odata instalat la nivelul creierului, acest agent transforma moleculele proteinice normale intr-un numar mare de copii infectioase, care sunt depozitate extracelular ca placi amiloide sau intracelular ca fibrile sau 'fasii' prionice (agregate de prioni).
Proteina structurala este o glicoproteina hidrofoba, cu o greutate moleculara de 28KDa, avand in compozitia sa 208-220 de aminoacizi, in functie de specie. Prezinta un capat NH2-terminal, o regiune centrala si un capat COOH-terminal. Domeniul NH2-terminal contine 80-100 de aminoacizi, o regiune alcatuita din cinci copii ale unei octarepetitii, existand cazuri in care acest numar este mai mare (de exemplu la bovine unde exista sase astfel de copii). Numeroase studii au aratat ca aceste repetitii au un grad inalt de conservare in seria animala, ceea ce inseamna ca indeplinesc un rol important in functionarea proteinei. La nivelul acestor repetitii se gaseste un situs de legare pentru ionii unor metale, in special pentru ionii de cupru si se presupune ca activitatea proteinei ar depinde de prezenta acestor ioni.
Acest domeniu se caracterizeaza printr-o plasticitate deosebita si prin prezenta unor structuri helicale, sugerand ca este implicat in conversia PrPc PrPSc.
Capatul COOH-terminal contine 120 de aminoacizi, o secventa semnal pentru atasarea unui glicofosfatidil inozitol care reprezinta "ancora" cu care PrP este prinsa de membrana celulara, si o regiune conservata, reprezentata de doua resturi de cisteina unite prin doua punti disulfidice. Proteina este procesata post-translational astfel incat are loc o rearanjare a unui numar variabil de aminoacizi la nivelul ambelor domenii terminale.
Forma patogena sau 'scrapie' (PrPSc) este identica din punct de vedere al compozitiei in aminoacizi cu proteina prionica normala, insa exista diferente marcante la nivelul structurii secundare si tertiare. Metodele biofizice, cum ar fi analizele RMN si dicroismul circular, au aratat ca exista diferente intre cele doua izoforme la nivelul structurii secundare, care constau in faptul ca PrPc este bogata in structuri a-helix, regiuni in care scheletul proteinic se rasuceste sub forma specifica a unei spirale, in timp ce PrPSc este bogata in legaturi b-pliate, in care proteina este desfasurata. Miezul proteinic al PrP contine 3 a-helixuri si o structura b-pliata scurta, restul moleculei prezentand o mobilitate crescuta. Originea diferentelor conformationale dintre cele doua forme este necunoscuta. Ele ar putea fi determinate fie de organizarea tertiara a monomerilor PrPSc, fie ar putea rezulta din structura cuaternara diferita a polimerilor PrPSc.
Functia PrPc este inca necunoscuta, insa si din acest punct de vedere exista o serie de ipoteze. Numeroase studii au fost realizate pentru a stabili rolul fiziologic al PrP la suprafata celulei si s-a constatat ca poate actiona ca un receptor celular pentru un ligand extracelular care nu a fost inca identificat. O alta ipoteza sugereaza ca PrP, la suprafata celulei, leaga ionii de cupru, fiind implicata in transportul si metabolizarea acestora. Prin izolarea unei octarepetitii din domeniul NH2-terminal, s-a observat ca prin legarea ionilor de cupru se formeaza o structura helicala ceea ce ar favoriza formarea proteinei anormale. Ionii metalelor tranzitionale redox-active cum sunt cupru si fierul, prezinta o importanta semnificativa in procesul neurodegenerarii, deoarece intra in structura unor enzime, iar deficienta lor poate duce la aparitia unor disfunctii la nivelul sistemului nervos central, generate de producerea de radicali liberi de oxigen si stress-ul oxidativ. Exista numeroase dovezi in sensul ca PrP are activitate superoxid dismutazica (SOD), concluzie rezultata din faptul ca celulele lipsite de PrP au o sensibilitate crescuta la stress-ul oxidativ datorita activitatii scazute sau chiar absente a SOD. Aceasta activitate este facilitata de legarea ionilor de cupru iar celulele in care proteina prionica este truncata si nu prezinta capatul NH2-terminal, prezinta o deficienta a SOD, deoarece ionii de cupru se leaga in special de acest capat.. In plus, ionii altor metale (in special ionii metalelor grele), pot media, prin fixarea lor la nivelul unui situs specific din proteina, neurotoxicitatea, favorizand formarea de agregate si, implicit, aparitia bolii. De asemenea, s-a sugerat ca aceasta proteina joaca un rol important in reglarea apoptozei, cu distrugerea nivelului celular normal al PrP in cursul infectiei, conducand ireversibil la moarte celulara.
Mecanismul formarii PrPSc
Formarea PrPSc este rezultatul procesului de conversie al structurilor a-helicale prezente in structura secundara a proteinei celulare, in structuri b-pliate caracteristice proteinei scrapie. Studii realizate cu peptide sintetice au sugerat ca o regiune din PrP, cuprinsa intre resturile de aminoacizi 108-121, poate determina formarea de legaturi b-pliate in izoforma patogena, PrPSc. Aceasta regiune pare a fi necesara pentru formarea PrPSc, deoarece deletia ei previne formarea PrPSc in timp ce deletia altor regiuni situate in amonte sau in aval de aceasta regiune, permite formarea acesteia. Procesul de conversie implica doar o schimbare in topologia proteinei, fara a avea vreun efect asupra compozitiei in amioacizi. In acest proces de conversie, s-a dovedit ca, din intreaga structura a proteinei celulare, un rol important il are, datorita plasticitatii sale crescute, domeniul NH2-terminal, care permite formarea unor structuri helicale. In ceea ce priveste identitatea factorului care determina acest proces, exista mai multe ipoteze, mai mult sau mai putin argumentate.
Dintre toate modele propuse, cel mai vehiculat este acela ca proteina nativa se gaseste in mod normal intr-un echilibru cu un ansamblu de de proteine care prezinta modificari conformationale minore - precursori monomerici ai PrPSc. Acesti precursori pot interactiona intre ei cu eficienta scazuta si reversibil, pana cand se formeaza un miez oligomeric infectios si stabil. Cand este atinsa o marime critica, precursorii monomerici ai PrPSc se pot aditiona la aceasta structura intr-un mod ireversibil, pentru a permite marirea particulei PrPSc. Particula infectioasa se poate apoi "reproduce" prin ruperea in particule mai mici si stabile. Cele patru etape (a) starea de echilibru dintre PrPc si PrPSc, b) interactia precursorilor, c) "reproducerea" si d) formarea unei particule stabile) ale acestui proces sunt schematizate in figura de mai jos.
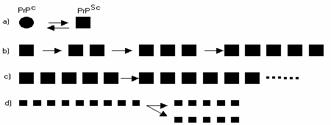
Figura 2. Etapele propagarii prionului.
Cand cantitatea de PrPSc depozitata atinge un nivel critic, se declanseaza o cascada auto-catalitica, care de obicei este ireversibila si care determina modificarea proteinelor normale nou sintetizate. In cazul maladiilor prionice, aceste depozite nu sunt distruse, ceea ce duce la acumularea lor in celule, determinand vacuolizarea si necroza acestora. S-a constatat ca in acest proces de conversie, PrPSc functioneaza ca matrita pentru formarea unor noi molecule PrPSc.
Graham S. Jackson si colab. (2000), au urmarit experimental schimbarea conformatiei proteinei PrPC in cursul procesului de conversie. Pentru aceasta, ei au incubat PrPc, marcata cu 32S, cu un exces de PrPSc si au observat ca se produce o proteina de novo, rezistenta la proteinaza K, pe care au desemnat-o PrPRES. Dupa digestia cu proteaza, se observa ca mobilitatea PrPRES in gel de elecrtoforeza SDS-PAGE este crescuta, ceea ce indica o scadere a masei moleculare, punand in evidenta faptul ca are loc o schimbare conformationala in proteina prionica celulara, in cursul procesului de conversie.
Unii autori sugereaza ca procesul de conversie poate fi indus si de factori fiziologici, cum ar fi valoarea pH-ului, cresterea temperaturii sau nivelul diferit de glicozilare al proteinei normale. De asemenea, exista unele supozitii, precum ca in procesul de conversie PrPc PrPSc ar fi implicati anumiti cofactori, unii dintre acestia apartinand familiei de proteine chaperone. Telling si colab. (1996), arata ca exista un alt factor auxiliar, numit "proteina X", care faciliteaza aceasta conversie prin legarea sa la un epitop discontinuu de la nivelul PrPc.
Se stie cu siguranta faptul ca formarea PrPSc este un proces posttranslational, care are loc doar dupa atasarea PrPc la membrana neuronala. Daude si colab. (1997) au aratat ca in cazul formelor ereditare, acest proces are loc chiar la nivelul reticulului endoplasmic, inainte ca proteina normala sa fie ancorata de membrana. Indivizii respectivi mostenesc direct forma mutanta, ceea ce explica absenta placilor amiloide la nivelul tesutului cerebral.
Au fost propuse variate ipoteze pentru a explica mecanismul care determina distrugerea celulelor neuronale si aspectul spongios al creierului. Acestea includ printre altele: efectul neurotoxic manifestat de o regiune a proteinei prionice, care cuprinde resturile de aminoacizi 106-126, precum si stress-ul oxidativ crescut la nivelul neuronilor ca urmare a depletiei PrPC, care se presupune ca functioneaza ca un antioxidant.
In cazul celor mai multe proteine globulare are loc o pliere a monomerilor care duce la formarea unei conformatii stabile. Cand plierea este incorecta sau cand apare o schimbare conformationala, ca urmare a substitutiei unui aminoacid, a unei modificari chimice, sau a unor factori necunoscuti, proteinele nu pot fi degradate si se agrega sub forma unor placi amiloide.
Maladiile prionice apar ca urmare a unor fenomene destabilizatoarea a structurii proteinei prionice normale, care o predispun la formarea de agregate. Interactia dintre PrPc si PrPSc ar putea fi facilitata de o mutatie, de legarea unui ligand sau a unei co-proteine.
Studii realizate pe soareci transgenici au aratat ca nici o deletie care are loc in amonte de aminoacidul din pozitia 106 a PrPC, nu determina aparitia unei maladii prionice, soarcele manifestand un fenotip normal, in timp ce orice deletie (mutatie punctiforma) aparuta in orice pozitie dupa situs-ul 106, conduce la ataxie severa precum si pierderea celulelor neuronale, simptome (si patologie) complet absente la soarecii ablated (soareci care prezinta o alogena cu deletie inaintea situs-ului 106). Aceste dovezi conduc la ipoteza ca PrP si o structura omoloaga sunt in competitie pentru un acelasi ligand.
Gena PrP
Proteina prionica normala este sintetizata pe baza informatiei genetice continuta intr-un fragment de ADN care se gaseste atat in celulele normale cat si in cele infectate, unde determina sinteza unei proteine normale, neprionice. Aceasta gena, la om, este localizata pe bratul scurt al cromozomului 20.
La mamifere, gena are trei exoni si un ORF (open reading frame) care este implicat in codificarea acestei proteine. Inactivarea sau deletia acestui ORF, in cele mai multe cazuri, are un efect nesemnificativ in dezvoltarea pre si neonatala a soarecilor, desi se stie ca expresia PrP este o conditie necesara, dar nu suficienta, pentru propagarea particulelor infectioase.
La hamsterul sirian, gena prezinta doi exoni separati de un intron de 10 kb. Exonul 1 codifica o portiune din secventa leader, situata la capatul 5', care va fi netranslata, in timp ce exonul 2 codifica pentru un ORF si secventa 3' netranslatata. La soarece, oi si sobolan, gena contine trei exoni, exonul trei fiind analog exonului doi de la hamster. Promotorii genei de la hamster si soarece contin regiuni bogate in G-C, repetate de un numar mare de ori si sunt de lipsiti de cutia TATA. Acesti nonameri G-C, reprezinta un motiv care poate functiona ca un sit de legare pentru factorul transcriptional Sp1. Analiza comparativa a cromozomului 20 la om (bratul scurt), pe care se gaseste gena, si a cromozomului 2 la soarece, a scos in evidenta faptul ca cele doua regiuni sunt omoloage, ceea ce inseamna ca aceasta gena este conservata in evolutie.
La pasari, gena contine un singur exon la nivelul caruia se gaseste un ORF, ceea ce elimina posibilitatea formarii PrPSc printr-un splicing alternativ.
Gena umana PRNP contine un singur ORF care codifica o proteina de 253 de aminoacizi. Aceasta proteina este procesata posttranslational, astfel incat are loc o rearanjare a unei secvente de 22 de aminoacizi de la capatul NH2-terminal. Acelasi lucru se intampla la capatul COOH, unde 23 de aminoacizi sunt rearanjati in cursul aditiei unui glicofosfatidil inozitol (GPI) care ancoreaza proteina de membrana celulara. Au fost studiate o multime de variante ale proteinei PrP, din punct de vedere al compozitiei chimice, la mai multe specii si s-a constatat ca doar secventa proteinica de la pui difera considerabil de cea umana. Din acestea se trage concluzia ca exista un grad inalt de conservare in seria animala si ca anumite functii s-au pastrat de-a lungul evolutiei.
Maladii prionice la om pot fi considerate afectiunile in care fiecare individ prezinta un progresiv declin subacut sau cronic al functiilor cognitive si motorii. De obicei, pacientii cu varsta cuprinsa intre 40-80 de ani, manifesta trasaturi clinice care permit stabilirea diagnosticului premorbid al maladiilor prionice, in special cei cu CJD.
Faptul ca 10% din aceste maladii sunt ereditare, duce la concluzia ca fondul genetic joaca un rol important. Mutatiile au loc la nivelul ORF-ului sau in regiunea codificatoare a genei, iar acest lucru a fost observat la toti pacientii cu maladii prionice mostenite. Ipoteza ca sindromul GSS ar putea fi transmis la maimute, a fost emisa prima data cand s-a presupus ca scrapia, CJD si alte maladii prionice ar fi determinate de un virus. Cand s-a descoperit ca mutatia la nivelul codonului 102 din gena umana, in urma careia prolina este inlocuita cu leucina este legata de aparitia maladiei GSS, s-a formulat ipoteza ca maladiile prionice sunt atat infectioase cat si genetice. Aceasta mutatie poate fi cauzata de deaminarea, in linia germinala, a unei insule CpG metilate, ceea ce duce la substitutia unei timine cu o citozina.
S-a constatat, atat la om cat si la animale, ca mutatiile au loc in anumite pozitii si ca fiecare din aceste mutatii este asociata cu un anumit fentip. De exemplu, mutatia la nivelul codonului 102 din gena umana este asociata cu inducerea GSS, mutatia aminoacidului din pozitia 129 a secventei proteinice predispune la CJD atat sporadic cat si ereditar, dubla mutatie din pozitiile 178 si 200 este asociata cu un alt fenotip GSS etc. De asemenea, s-a observat ca un fenotip de CJD poate fi cauzat de insertiile unor octarepetitii. Aditia a 2, 4, 5, 6, 7, 8 si chiar 9 repetitii la cele cinci prezente in mod normal, au fost identificate la indivizi care prezentau CJD ereditar; deletia unei asemenea repetitii nu este corelata cu aparitia bolilor neurodegenerative. Mutatia punctiforma de la nivelul codonului 178 este corelata cu instalarea FFI (insomina familiala fatala).
Analizele de genetica moleculara au scos in evidenta polimorfismele de la nivelul genei PrP. Polimorfismul din pozitia 129 pare a fi capabil sa influenteze expresia maladiilor prionice nu numai in formele ereditare dar si in cele sporadice sau infectioase.
Formele sporadice se refera in special la cazurile de CJD si posibil, (intr-o mica masura), la cele de GSS. In cazul acestor pacienti nu s-au identificat mutatii la nivelul genei. Modul de aparitie a maladiei la acesti indivizi nu este cunoscut; se presupune ca ar putea avea loc o transmitere a factorului etiologic pe orizontala (intre indivizii aceleiasi specii sau apartinand la doua specii diferite), ca urmare a unor mutatii somatice la nivelul ORF-ului genei PrP sau prin conversia spontana a PrPc in PrPSc. Toate mutatiile punctiforme identificate la nivelul genei PrP, au loc fie in secvente din interiorul, fie din apropierea unei regiuni din structura secundara a PrP, si, in felul acesta, determina destabilizarea structurii proteinei prionice.
Desi cunoscute de sute de ani, maladiile prionice reprezinta inca un domeniu putin cunoscut din punct de vedere biologic si medical fiind necesare, inca, numeroase studii si cercetari pentru stabilirea cu exactitate a cauzelor si a mecanismului care determina aparitia acestor boli. Faptul ca exista numeroase variante ale acestor boli, si ca pot fi de natura diferita, reprezinta o bariera in realizarea acestui obiectiv.
Ipotezele formulate pentru descifrarea mecanismului acestor boli arata ca acesta este comun atat la om cat si la animale si ca la baza lui se afla procesul de conversie a PrPc in PrPSc.
Acest proces de conversie poate fi indus de o serie de factori, atat endogeni cat si exogeni, si consta in schimbarea conformationala a proteinei normale (conversia structurilor a-helix in b-pliate) fara a afecta in nici un fel compozitia chimica a acestor proteine.
Un rol important in acest proces il au mutatiile de la nivelul genei care codifica proteina normala; acestea au loc in pozitii fixe si sunt corelate cu anumite fenotipuri.
Desi PrP este implicata in procesul neurodegenerativ, ramane neclar daca neurotoxicitatea PrPSc sau pierderea functiei PrPc (de exemplu activitatea superoxid dismutazica), este determinantul major al manifestarilor clinice.
Exista multe intrebari la care nu s-au gasit inca raspunsuri in ceea ce priveste relatia dintre structura si functia acestei proteine prionice.
1) Brockes PJ., - Topics in prion cell biology, Current Opinion in Neurobiology, 1999, 571-577.
2) Daggett V., - Structure-function aspects of prion proteins, Current Opinion in Biotechnology, 1998, 9: 359-365.
3) Daude N, Lehmann S, Harris DA, - Identification of intermediate steps in the conversion of a mutant prion protein to a scrapie-like form in cultured cell, J Biol. Chem. 1997, 272: 11604-11612.
4) Foster J., Hunter N., - Transmissible spongiform encephalopathies: transmission, mechanism of disease, and persistence, Current Opinion in Microbiology, 1998, 1: 442-447.
5) Hope J., - Prions and neurodegenerative diseases, Current Opinion in Genetics & Development, 2000, 10: 568-574.
6) Gee H., - Molecular Evolution of Prions, Nature, 1996.
7) Graham SJ., Collinge J., - Prion disease-the propagation of infectious protein topologies, Microbes and Infection, 2000, 2: 1445-1449.
8) Graham SJ, Clarke AR., - Mammalian prion proteins, Current Opinion in Structural Biology, 2000, 10: 69-74.
9) Markus G., Aguzzi A., - Peripheral pathogenesis of prion diseases, Microbes and Infection, 2000, 2: 613-619.
10) Prusiner SB., Scott MR., - Genetics of Prions, Annu. Rev. Genet., 1997, 31: 139-175.
11) Rezaie P, Lantos PL, - Microglia and the pathogenesis of spongiform encephalopathies, Brain Research Reviews, 2001, 35:55-72.
12) Telling GC, Parchi P, DeArmond SJ, Cortelli P, Montagna P, Gabizon R, Prusiner SB, - Evidence for the conformational of the pathologic isoform of the prion protein enciphering and propagating prion diversity, Science, 1996, 274, 2079-2082.
13) Wadsworth JDF, Graham SJ, Collinge J.,Hill AF., - Molecular biology of prion propagation, Current Opinion in Genetics & Development, 1999, 9: 338-345.
14) Wickner RB, Edskes HK, Maddelein ML - Prions: Portable prion domains, Current Biology,. 2000,10: 335-337.